TCS Tutorial A quick start to evaluate input alignment by Transitive Consistency Score (TCS)
Description
The web server allows you to measure alignment confidence based on T-Coffee framework, TCS [1] and its downstream application for phylogenetic reconstruction based on PhyML [6].
TCS is an alignment evaluation score that makes it possible to identify in an MSA the most correct positions. It has been shown that these positions are the most likely to be structurally correct and also the most informative when estimating phylogenetic trees.
The TCS evaluation and filtering procedure is implemented in the TCS web server, as well as T-Coffee package. It can be used to evaluate and filter any third party multiple sequence alignment (including T-Coffee of course!).
Data input
Default model
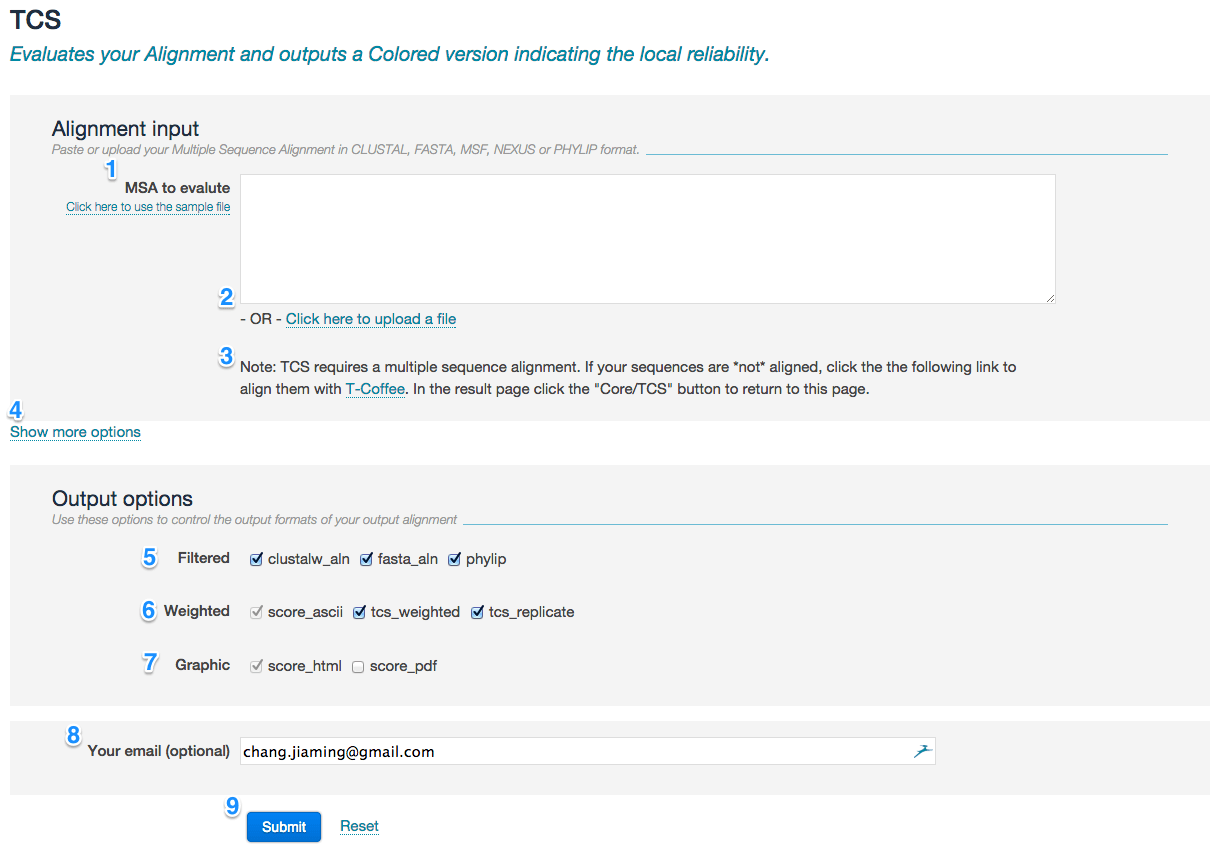
- Enter the MSA in the input field (marked with 1 in the picture below) or alternatively provide them using the Upload link (2).
- MSA must be in CLUSTAL, FASTA, MSF, NEXUS or PHYLIP format. The limit of MSA size is 1000 sequences and sequence length is 5000.
- TCS requires a MSA input. If your sequences are NOT aligned, you can run any T-Coffee method first (i.e., the link for regular T-Coffee) and then click a blue button ‘Core/TCS’ in the ‘Send results’ section of the result page, which will automatically initialize pre-filled TCS service with T-Coffee (3).
- You may access to TCS advanced settings by clicking the link Show more options (4). Please refer to the “advance model” section for detailed information.
-
Use these options to control the output formats of the evaluation:
- Filtered (5): Specify alignment output format after TCS-based filtering procedure. It is suggested to select "phylip" if you want to perform downstream phylogenetic reconstruction. Default: clustalw_aln, fasta_aln, phylip.
- Enriched alignment (6):
- score_ascii Outputs a TCS evaluation score in plain text format
- tcs_weighted Outputs phylip format with duplicated columns according to their TCS score
- tcs_replicate Generates 100 phylip replicates, with columns drawn according to their TCS score
- Graphic (7):
- score_html Outputs a TCS evaluation score in html format
- score_pdf Will transfer score_html into pdf format
- Provide your email address (8) if you want to be notified when the job complete.
- Press the button submit (9) to submit your request.
Please note that the computation can take some minutes to be fulfilled. You don't need to keep your browser open to wait for the result. You can close it or navigate away to another page.
Use the History link, in top-bar menu to get access to the computation result at any moment.
You can also retrieve your results using the permanent URL http://tcoffee.crg.cat/apps/tcoffee/result?rid=jobid, where jobid is the ID given at the time of submission.
Advance model
Filter options:
- You can use these options to control the filtering intensity of your output alignment and whether you want the filtering to remove columns (recommended) or residues (1).
- All columns with a TCS score lower than minimum or larger than maximum are turned into gaps (2). Default: minimum = 4, maximum = 9
- Remove empty columns which contains all gaps after the above filtering (3). Default: yes
-
Your alignment is evaluated by comparison against a collection of alignments. This collection is named a Library.
This section lets you control the computation of the library. Different libraries give different score results (4):
- Mproba_pair It is the default mode of T-Coffee that runs a pair-HMM to populate the library with residue pairs having the best posterior probabilities.
- Mkalign_msa / Mmafft_msa / Mmuscle_msa This mode runs a series of fast multiple aligners. It is very fast and used by ENSEMBL Compara. Please use this mode if your alignment contains more than 100 sequences.
- Mlalign_id_pair / Mclustalw_pair This mode runs the original default T-Coffee that was combining local and global alignments.
- Mfast_pair / Mslow_pair

Output interpretation
The result page contains the following sections:
- Citation The related articles when citing the TCS.
- MSA The MSA is colored according to T-Coffee TCS scheme. Dark pink bits are very reliable, while blue and green bits are unreliable. Color has been redesigned to be visualized for color blind people.
- Result files
All files can be downloaded as a single zip file or copied to the user’s Dropbox account.
- The Multiple Alignment subsection provides a filtered version of the original MSAs in CLUSTAL, FASTA and Phylip formats that can be used for phylogenetic reconstruction or homology modeling.
- score_ascii The plain text format indicating the ResidueTCS, ColumnTCS, SequenceTCS and AlignmentTCS scores, respectively.
- score_html The html format.
- tcs_weighted In which all columns are duplicated a number of times equal to their ColumnTCS.
- tcs_replicate That contains a 100 bootstrap replicates of the original MSA with column randomly selected weighed by their ColumnTCS.
-
Phylogenetic reconstruction
Estimate a phylogenetic tree from selected MSAs using the PhyML web server.
- Send Filtered MSA to PhyML: automatically initial a PhyML job with the filtered MSA.
- Send Weighted MSA to PhyML: automatically initial a PhyML job with the tcs_weighted.
- Send Bootstrap MSA to PhyML: automatically initial a PhyML job with the tcs_replicate.
- Send results Forward this result to other online tools.
- Info Some information related to the submitted job (running time, date, etc). You may want to use the full featured TCS through command line mode online.
- Replay this link allows the user to re-run the job while modifying input options or data.
- Feedback To share your alignment experience in the social network.

Supplemental Data
-
TCS method
Chang JM, Di Tommaso P, Notredame C.
TCS: a new multiple sequence alignment reliability measure to estimate alignment accuracy and improve phylogenetic tree reconstruction.
Molecular biology and evolution 31, 1625–37 (2014). (PMID: 24694831) -
TCS web server
Chang, J.-M. M., Di Tommaso, P., Lefort, V., Gascuel, O. & Notredame, C.
TCS: a web server for multiple sequence alignment evaluation and phylogenetic reconstruction.
Nucleic Acids Res. (2015). doi:10.1093/nar/gkv310 (PMID: 25855806) -
Original CORE publication
Notredame and Abergel, in Bioinformatics and Genomes: Current Perspectives. M. Andrade, Horizon Scientific Press: pp 30-50 - TCS project site
- TCS standalone usage
- PhyML web site